|
 |
 |
|
|||||||||||||||||||||||
 |
 |
|||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||
|
| |||||||||||||||||||||||||||||||||||||
|



 |
Налоксон и налтрексон нашли широкое применение в медицине. Они являются конкурентными антагонистами опиоидных рецепторов. В исследованиях на мутантных рецепторах показано, что многие аминокислотные остатки трансмембранных доменов играют важную роль в связывании налоксона и налтрексона. Показано, что даже отдельные мутации могут существенно изменять сродство антагонистов к рецепторам, а в некоторых случаях - обеспечивают появление у них свойств агонистов. Длительные воздействия налоксоном и налтрексоном сопровождаются возрастанием плотности опиоидных рецепторов и повышением чувствительности к агонистам. Обсуждаются подобная суперчувствительность и возможность передозировок у героиновых наркоманов после длительного приема налтрексона. А. Головко* , С. Головко, Л. Леонтьева**, О. Романенко, Д. Коноплин А. Головко* , С. Головко, Л. Леонтьева**, О. Романенко, Д. Коноплин Молекулярные аспекты фармакологической активности налтрексона и налоксона
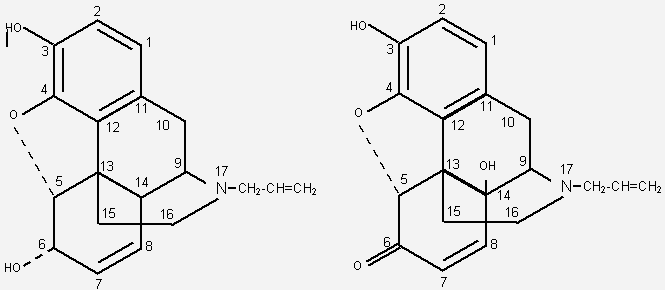
ВведениеИзучение фармакологических свойств антагонистов опиоидных рецепторов получило развитие в начале 20 века, когда была продемонстрирована способность N-аллилноркодеина ослаблять угнетающее действие морфина и героина на дыхательный центр [58]. В 40-х годах в лечебную практику стал внедряться налорфин (рис. 1). Долгое время он оставался основным антидотом при острых отравлениях морфином [27]. Налоксон, синтезированный в 1960 г., значительно превосходил налорфин по эффективности и по терапевтической широте [33, 52]. К его недостаткам следует отнести короткий период полувыведения и отсутствие лекарственных форм для перорального приема. В значительно большей степени перечисленным требованиям отвечает налтрексон (синтезирован в 1963 г.) [52]. Рис.1. Химические структуры антагонистов опиоидных рецепторов.
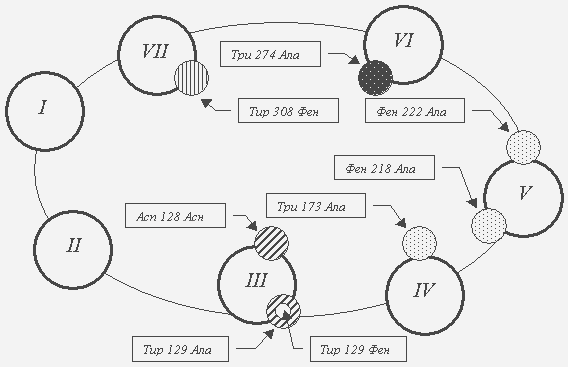
Налтрексон Антагонистические свойства налоксона и налтрексона первоначально выявлены на уровне физиологических и поведенческих реакций. Оба препарата эффективно устраняли фармакологические ответы на введение опиатов/опиоидов (анальгезия, седативное действие, угнетение дыхательного центра и т.д.) [20, 23, 33, 83]. Данное свойство стало основой высокой антидотной активности налоксона при острых отравлениях агонистами опиоидных рецепторов. При совместном введении налоксона или налтрексона с наркотическими анальгетиками удавалось предупреждать развитие толерантности и абстинентного синдрома [23]. Наиболее ярким доказательством антагонистической активности препаратов считают их способность ускорять и усиливать симптомы лишения наркотика (precipitated withdrawal) [48]. По этому показателю налтрексон значительно превосходит налоксон [8, 9]. В исследованиях на опиатных наркоманах-добровольцах доказана способность антагонистов противодействовать эйфоригенным эффектам героина и морфина [18, 62]. Это обеспечило выраженную противорецидивную активность налтрексона у наркоманов, прошедших начальный этап абстиненции. Рис. 2. Влияние замен аминокислотных остатков на сродство дельта-рецептора к налоксону [6].
Примечания: 1. Круги с римскими цифрами - трансмембранные домены; маленькие круги - места замен аминокислот; характер узора внутри маленького круга соответствует определенной степени снижения аффинитета мутантной формы рецептора по сравнению с "диким" типом. 2. Снижение аффинитета рецепторов (кратность:
3. Характер мутации отражен в прямоугольнике: “Тир 129 Ала” означает, что тирозин в положении 129 заменена на аланин. 4. Сокращения названий аминокислот: Фен – фенилаланин, Асп – аспартат, Асн – аспарагин, Три – триптофан. Нейрохимическую основу фармакологической активности антагонистов составляет их способность связываться с опиоидными рецепторами. Специфическое связывание налоксона и налтрексона доказано в радиолигандных исследованиях [60, 76], с помощью радиоаутографии [50] и позитронно-эмиссионной томографии [37, 71]. Следует учитывать, что способность к связыванию с опиоидными рецепторами дает основание причислять налоксон и налтрексон к специфическим лигандам. Доказательством же фармакологической активности на нейрохимическом уровне может рассматриваться способность модулировать системы вторичных и третичных мессенджеров, экспрессию генов, функциональную активность неопиоидергических нейромедиаторных систем [21, 32, 39, 66]. 1. Молекулярные основы связывания антагонистовНаиболее ранние радиолигандные исследования с использованием [3Н]-налоксона и [3Н]-налтрексона выполнены в начале 70-х годов 20 века [54-56, 65]. В последующие годы высокий аффинитет налоксона и налтрексона к опиоидным рецепторам подтвержден с помощью радиоаутографии и позитронно-эмиссионной томографии [37, 50, 71]. Оба препарата обладают сродством ко всем трем подтипам опиоидных рецепторов (мю, дельта и каппа). Наиболее эффективно они связываются с мю-рецепторами. Так, анализ результатов специфического связывания с [3Н]-налтрексона с синаптическими мембранами коры больших полушарий крыс выявил 2 сайта рецепторов – высоко- и низкоаффинный. Первый соответствовал мю-, а второй – дельта-рецепторам. Связывание с каппа-подтипом было минимальным [61]. Сродство налтрексона к мю- и дельта-рецепторам существенно превосходит соответствующий показатель для налоксона. Так, константа диссоциации (Kd - концентрация лиганда, при которой насыщается половина рецепторов; Kd обратно пропорциональна сродству лиганда к рецепторам) при использовании [3Н]-налтрексона составляла менее 1нМ [56, 76], а для [3Н]-налоксона, как правило - более 1 нМ [43, 60]. Сходные данные получены при оценке сродства с помощью вычисления константы ингибирования антагонистами специфического связывания селективных радиолигандов мю-, дельта- и каппа-рецепторов [10, 28, 46]. Для понимания механизмов рецептирования антагонистов целесообразно кратко напомнить о структуре опиоидных рецепторов. Опиоидные нервные окончания принадлежат к семейству метаботропных рецепторов, ассоциированных с G-белками. Передача информации на постсинаптические структуры осуществляется посредством модуляции различных систем вторичных мессенджеров: аденилатциклазы, каналов кальция и натрия, фосфоинозитидного каскада [26, 36, 41]. Молекула рецептора включает внеклеточный NH2-участок. Далее следуют семь трансмембранных доменов (ТМ I – ТМ VII), между которыми расположены три внеклеточные и три внутриклеточные петли. Завершает полипептидную цепь внутриклеточная СОО-терминаль [5, 6, 26, 56]. Предполагается, что молекула рецептора свернута в спираль, а ее трансмембранные домены тесно взаимодействуют между собой [5, 6, 26, 47, 56]. Внеклеточный домен имеет несколько мест для гликозилирования по остаткам аминокислоты аспарагин. Между первой и второй внеклеточными петлями выявлена дисульфидная связь между остатками цистеинов [82]. Зона рецептирования лигандов опиоидного рецептора условно делится на участки селективности и карман связывания. Первые расположены преимущественно выше наружной поверхности мембраны и сформированы аминокислотными остатками внеклеточных петель и верхушек трансмембранных доменов [47, 49]. Карман находится ниже наружной поверхности мембраны. Он ограничен спиральными петлями трансмембранных доменов [5, 6]. В рецептирование лигандов пептидной природы вовлечены как участки селективности, так и карман связывания. Алкалоиды и их дериваты связываются преимущественно в области кармана. При этом заряженный азот молекулы лиганда способен взаимодействовать с остатками ароматических аминокислот полипептидной цепи [5, 6, 45, 47]. Углублению теоретических представлений о механизмах рецептирования лигандов опиоидных рецепторов способствовало внедрение методов клонирования. В частности, сведения об участках связывания получены посредством создания химерных конструкций, когда полипептид формировался из фрагментов, принадлежащих рецепторам различных типов: мю/дельта, дельта/мю, дельта/каппа и т.д. [17]. Подобный подход позволил говорить о конкретном участии внеклеточных петель и трансмембранных доменов в связывании агонистов и антагонистов мю-, дельта- и каппа-рецепторами [41]. Исследования с химерными рецепторами позволили выявить участки, ответственные за формирование селективности к различным лигандам. Но этот подход не обеспечивал возможности определить ключевые аминокислоты. Поэтому следующим шагом стало изучение вклада отдельных аминокислотных остатков посредством клонирования мутантных рецепторов, в которых заменялась одна или несколько аминокислот. Оценивались функциональные характеристики рецептора (по параметрам связывания радиолигандов, по активности систем трансдукции), а также осуществлялось компьютерное моделирование участка рецептирования [6]. Наиболее подробно данная проблема изучена в отношении связывания агонистов пептидной и непептидной природы [10, 60, 74]. Однако накоплен достаточный материал и для понимания механизмов рецептирования антагонистов (табл.). Таблица Влияние мутаций различных аминокислот на состояние опиоидных рецепторов
* Осуществлена замена аспартат-128 на аланин в третьем трансмембранном домене Как следует из данных таблицы, специфическое связывание налоксона и налтрексона изменяется (чаще в сторону снижения) при замене аминокислот во втором – седьмом трансмембранных доменах. В настоящем обзоре не приводятся результаты изучения связывания агонистов с рецепторами-мутантами. Между тем, анализ многочисленных работ свидетельствует, что в условиях сайт-направленного мутагенеза рецептирование агонистов сопровождалось более драматическими нарушениями в сравнении со связыванием антагонистов. Особенно это характерно для агонистов пептидной природы [12, 75]. K.Befort и др. [6] оценивали роль различных аминокислот трансмембранных доменов в рецептировании налоксона мутантными дельта-рецепторами. Значимость ароматических аминокислот определяли посредством следующих мутаций: Триптофан 173 Аланин (ТМ IV) * ; Фениилаланин 218 Аланин и Фенилаланин 222 Аланин (ТМ V). О роли гидроксильных групп – посредством замен Тирозин 129 Фенилаланин (ТМ III) и Тирозин 308 Фенилаланин (ТМ VII). Наконец, с помощью замены тирозина в положении 129 (TM III) на аланин судили об участии ароматического кольца в формировании мест специфического связывания антагониста (рис. 2). Подтверждена важная роль ароматических аминокислот в рецептировании налоксона. Так, замена тирозина-129 на другую ароматическую аминокислоту, фенилаланин, мало влияла на связывание антагониста, тогда как замещение на аланин сопровождалось снижением сродства в десятки раз. К важным (ключевым) аминокислотам кармана связывания относят глутаминовую и аспарагиновую кислоты, гистидин [5, 67, 69]. Например, J. G. Li и др. [45] установили, что в связывании налтрексона важную роль играет отрицательно заряженная карбоксильная группа аспартата-147 (ТМ III) мю-рецептора. По-видимому, происходит образование ионной связи между карбоксилом аспартата-147 и протонированным азотом-17 налтрексона. Замена аспартата-147 на аспарагин или на аланин, которые не имеют подобной группы, резко понижала аффинитет рецептора к антагонисту. Мутанты с глутаматом-147 (имеется карбоксил в положении 147) мало отличались от рецепторов “дикого” типа. Авторы представили еще одно доказательство своей концепции. Аналог налтрексона, не подвергающийся протонированию, обладал очень низким сродством к рецептору, а его антагонистическая активность в сравнении с налтрексоном была слабее в 100 раз. Предполагается, что в образовании ионной связи с налоксоном и налтрексоном могут принимать участие и карбоксилы глутаминовой кислоты. Структура кармана связывания опиоидного рецептора модулируется аминокислотными остатками, расположенными на некотором удалении от него. К примеру, замена лизина-108 в первой внеклеточной петле на аспарагин сопровождалась многократным снижением аффинитета дельта-рецептора к налоксону. Первая экстрацеллюлярная петля, как известно, не участвует в формировании кармана связывания, с которым взаимодействует антагонист. Здесь речь идет о серьезных внутримолекулярных перестройках [49]. 2. Влияние антагонистов на системы вторичных мессенджеровАрхитектоника рецепторного комплекса настолько детерминирована, что даже одна мутация может привести к серьезным изменениям параметров взаимодействия лиганда и рецептора. В некоторых исследованиях налоксон и налтрексон на рецепторах-мутантах проявляли свойства частичных или полных агонистов. В работах [67, 68] мю-рецепторы экспрессировали в ооцитах Xenopus laevis, после чего оценивали калиевые токи, регулируемые G-белком. На рецепторах “дикого” типа налоксон и налтрексон не изменяли калиевые токи, т.е. проявляли свойства антагонистов. В экспериментах с мутантами Гистидин 297 Аспарагин и Гистидин 297 Глутамин выявлялась агонистическая активность налоксона и налтрексона. Авторы полагают, что замена гистидина-297 (TM VI) на аспарагин или глутамин сопровождается изменением взаимодействия трансмембранных доменов с третьей внутриклеточной петлей, которая, в свою очередь, тесно связана с G-белком. Появление агонистической активности налоксона и налтрексона в отношении калиевых каналов, сопряженных с G-белком, также продемонстрировано на мутантных формах мю-рецептора Серин 196 Лейцин (TM IV) и дельта-рецептора Серин 177 Лейцин (TM IV) [19]. Как известно, мю- и дельта-агонисты угнетают базальную и стимулированную активность аденилатциклазы. Налоксон и налтрексон противодействуют этим эффектам, но самостоятельно не влияют на фермент [4]. На клетках яичника китайских хомячков, экспрессирующих мутанты мю-рецептора Серин 196 Лейцин или дельта-рецептора Серин 177 Лейцин, показана агонистическая активность налоксона и налтрексона. Это выражалось в появлении у блокаторов способности угнетать аденилатциклазу, стимулируемую форсколином [19]. Сходным образом проявлял себя налоксон и на дельта-мутанте Аспартат 128 Лизин [15]. Налоксон и налтрексон способны проявлять агонистическую активность не только на биологических системах, содержащих мутантаные опиоидные рецепторы. Уже в ранних клинических испытаниях налтрексона, выполненных в 70-80 гг., отмечена способность препарата вызывать миоз, снижение частоты дыхания и другие симптомы. Подобные эффекты свойственны опиоидным агонистам (см. обзоры [13, 33]). В последующие годы с использованием некоторых нейрохимических и электрофизиологических моделей удалось продемонстрировать агонистическую активность налоксона и налтрексона. Так, налоксон угнетал аденилатциклазу, стимулированную форсколином. Биологической системой в данном случае стали клетки яичника китайских хомячков, коэкспрессирующие аденилатциклазу и мю- или каппа-опиоидные рецепторы. Этот эффект был слабее в сравнении с действием “классических” опиоидных агонистов, но само его присутствие позволило авторам говорить о частичной агонистической активности налоксона [31]. Установлена способность налоксона изменять частоту сердечных сокращений и двигательную активность плодов свиньи в период поздней беременности. Подобные эффекты напоминали действие морфина в сходных условиях эксперимента [20]. 3. Реакции биологических систем на хроническое воздействие антагонистамиХроническое воздействие налоксоном и налтрексоном сопровождается повышением плотности опиоидных рецепторов в головном мозге экспериментальных животных (опыты ex vivo) и в клеточных культурах (опыты in vitro) [55, 78, 81]. Подобное явление получило название up-regulation. В некоторых случаях данное состояние сопровождалось повышением чувствительности биологической системы к фармакологическим эффектам опиатов/опиоидов [34, 77, 78]. Агонисты оказывают противоположное действие на число рецепторов – down-regulation [25]. Причем, если состояние up-regulation при длительных экспозициях к антагонистам стабильно достигается в большинстве биологических моделей, то подавление плотности рецепторов с помощью агонистов наиболее эффективно в опытах in vitro [81]. Среди нейрохимиков и наркологов сформировалась точка зрения о том, что увеличение числа опиоидных рецепторов является одним из проявлений длительного воздействия налоксоном и налтрексоном [77, 78, 84]. Однако есть исследования, показывающие, что на начальном этапе взаимодействия антагонистов с нервными окончаниями может развиваться состояние down-regulation. Такой феномен свидетельствует о проявлении налоксоном и налтрексоном свойств агонистов [7, 31]. В опытах in vitro показана способность налоксона действовать на возбужденные опиоидные рецепторы в качестве обратного агониста. Это означает, что антагонист обладает внутренней негативной эффективностью и стабилизирует рецептор в неактивном состоянии [24]. Итак, есть основания полагать, что налоксон и налтрексон, хотя и относятся к антагонистам, способны изменять конформационное состояние опиоидных рецепторов, проявляя тем самым свойства частичных агонистов. Практикующим наркологам необходимо знать: насколько важны приведенные эффекты для реализации фармакологической активности антагонистов. Например, при использовании налоксона и налтрексона в качестве антидотов при острых отравлениях опиатами/опиоидами агонистическая активность может рассматриваться как нежелательная. Для проведения заместительной терапии рассматриваемые препараты не используются из-за явного преобладания антагонистического профиля. Однако включение налоксона в схему противорецидивного лечения частичным агонистом бупренорфином резко повысило качество терапии [40]. Появились сообщения о способности налоксона и налтрексона в низких дозах (концентрациях) достоверно усиливать антиноцицептивную активность морфина и опиоидных анальгетиков (опыты in vitro и in vivo). Это подтверждает ранее высказанное предположение о влиянии антагонистов на конформационное состояние опиоидных рецепторов. Включение блокаторов в схемы лечения болевого синдрома не только повышает эффективность терапии, но и противодействует развитию толерантности и зависимости [23]. В практике нарколога антагонисты применяются при лечении опиатной наркомании и зависимости от алкоголя. В частности, их способность ускорять опиатный абстинентный синдром (precipitated withdrawal) используется в процессе ультрабыстрой опиатной детоксикации [53, 63]. В схемах противорецидивной терапии алкоголизма и опиатной наркомании применяют налтрексон [30, 42]. Долгое время основной лекарственной формой для профилактики рецидивов употребления опиатов/опиоидов или алкоголя служили таблетки или капсулы налтрексона. Однако в 70-х гг. начались исследования по созданию пролонгированных форм налтрексона, пригодных для имплантации под кожу [29]. Была разработана и успешно использована в клинических испытаниях суспензия микрокапсул [42], гелеобразные препараты налтрексона [30]. Изучается возможность создания пролонгированных таблетированных форм [2]. По мере внедрения налтрексона в клиническую практику накапливались сведения о побочных эффектах. К ним относили гепатотоксичность [57, 72], расстройства функции желудочно-кишечного тракта (тошнота, диарея), кожную сыпь, головную боль, ознобы, тревогу, дисфорию [13, 22, 33]. К настоящему времени появились сообщения о таких осложнениях, как кома [1], рабдомиолиз [16], отек легких [35]. Высказывалась точка зрения о том, что длительный прием антагониста может сопровождаться повышением чувствительности пациентов к токсическому действию опиатов/опиоидов [3, 78]. Рассмотрим подробнее эту позицию. Ранее упоминалось, что хроническое воздействие налоксоном или налтрексоном индуцирует состояние up-regulation, т.е. увеличение числа опиоидных рецепторов [55, 78]. Первые работы, продемонстрировавшие повышение числа опиоидных рецепторов в головном мозге на фоне хронического введения антагонистов, выполнены в начале 70-х гг. [38, 55]. При этом речь шла о возрастании суммарной плотности опиоидных нервных окончаний. Значительно позже было выяснено, что явление up-regulation распространяется на все подтипы рецепторов: мю, дельта и каппа [14, 64, 80]. Однако в исследовании [70] изменения каппа-подтипа в подобных условиях эксперимента не выявлены. В ряде работ показано, что на фоне состояния up-regulation возрастает чувствительность экспериментальных животных к фармакологическим эффектам опиатов/опиоидов [34, 84]. Вполне понятна настороженность некоторых исследователей в отношении возможности повышения токсичности опиоидных наркотиков у пациентов, окончивших курс лечения налтрексоном [3, 78]. Данный вопрос требует дальнейшего изучения [73]. Многочисленные эксперименты на животных свидетельствуют, что такой вариант событий вполне вероятен. Хроническое воздействие блокаторами сопровождается повышением чувствительности животных к фармакологическому действию морфина и других опиоидных агонистов. Параллельно выявляется возрастание плотности опиоидных рецепторов в головном мозге [77, 78]. Например, в исследовании [70] после длительной инфузии налтрексона в мозге крыс выявлялось возрастание плотности мю-опиоидных рецепторов. Через 6 суток данный показатель возвращался к контрольным значениям. Сходным образом изменялась чувствительность животных к анальгетическому действию морфина (возрастание после отмены блокатора и снижение до контрольного уровня на 6-й день). С этими результатами согласуются данные D.Levesque и S.G.Holtzman [44]. В их экспериментах крысы получали хронически налоксон. Возрастание анальгетической активности морфина выявлялось через 24 часа, но не к исходу 6-х суток после отмены антагониста. Окончательное заключение о возрастании числа передозировок у героиновых наркоманов, получающих налтрексон в качестве противорецидивной терапии, можно будет сделать только после проведения широкомасштабных исследований. Вероятно, такая информация будет получена после обследования пациентов, которым осуществлялась имплантация блокатора. Подобная форма использования налтрексона в рамках противорецидивного лечения опиатной наркомании все шире внедряется в ряде стран [30]. Было бы заманчиво считать состояние up-regulation нейрохимическим коррелятом повышения фармакологической активности опиатов/опиоидов в условиях хронического воздействия антагонистами. Однако анализ ряда работ позволяет усомниться в справедливости такой постановки вопроса. Например, при одновременном хроническом воздействии на грызунов налтрексоном и опиоидными агонистами не удалось добиться повышения чувствительности животных к наркотикам в конце эксперимента. При этом в мозге выявлялось стойкое повышение плотности опиоидных рецепторов [79]. J.L.Neisewander и др. [51] оценивали анальгетическую активность морфина и его способность влиять на спонтанную двигательную активность крыс разного возраста после длительной инфузии налтрексона. В параллельной серии экспериментов с помощью радиолигандного анализа оценивали плотность опиоидных рецепторов в переднем стриатуме животных. Выяснено, что после длительного воздействия антагонистом фармакологическая активность морфина повышалась только у молодых и взрослых крыс. У старых животных не выявлялось различий в контрольной и опытной группах. Вместе с тем, у грызунов всех возрастов длительная инфузия налтрексона сопровождалась стойким возрастанием плотности опиоидных нервных окончаний. Можно заключить, что между увеличением фармакологической активности опиатов/опиоидов и явлением up-regulation при хроническом воздействии антагонистами далеко не всегда удается провести четкие корреляционные связи. Таким образом, налоксон и налтрексон представляются важными лекарственными препаратами в современной наркологии. Достигнуты определенные успехи в обосновании их фармакологической активности. Этому способствовали многочисленные клинические испытания антагонистов, исследования в области нейробиологии и нейрохимии опиоидергических нейромедиаторных систем. Сформирована серьезная база клинических и экспериментальных данных, позволяющая оптимизировать лечебное использование налоксона и налтрексона и наметить направления поиска новых антагонистов опиоидных рецепторов. Литература
Региональный лечебно-диагностический медицинский центр "Бехтерев", 198096 Санкт-Петербург, ул. Корабельная д.6, Тел. Сведения об авторах Головко Александр Иванович. Заведующий стационарным отделением регионального лечебно-диагностического медицинского центра "Бехтерев", доктор медицинских наук профессор. 195027, Санкт-Петербург, Большеохтинский пр., дом 31, кв.5; д.т. 227-20-22, (отв. за переписку), сл. тел. 183-93-57, 183-92-75, 324-04-18. E-mail: prgolovko@mail.ru Головко Cергей Иванович. Старший научный сотрудник Центра, кандидат медицинских наук. 197348, Санкт-Петербург, Богатырский пр., дом 4, кв.724; д.т. -393-11-18, сл. тел. 183-93-57, 183-92-75, 324-04-18, 393-85-09. Леонтьева Любовь Владимировна. Ассистент-исследователь Университета шт.Западная Виржиния, Box 9151, Morgantown, WV 26505-9151, U.S.A., тел. (304) 293-15-28, факс (304) 293-02-65, E-mail: Luba105@hotmail.com. Романенко Олег Иванович. Ординатор стационарного отделения Центра, кандидат медицинских наук. 198005, Санкт-Петербург, 4-я Красноармейская ул., дом 2/35, кв. 45; д.т. 316-40-79, сл. тел. 183-93-57, 183-92-75, 324-04-18. Коноплин Дмитрий Алексеевич. Ординатор стационарного отделения Центра. 193177, Санкт-Петербург, ул. Караваевская, дом 22, кв. 15; д.т. 107-39-42, сл. тел. 183-93-57, 183-92-75, 324-04-18. Abstract A.I.Golovko*, S.I.Golovko, L.V.Leontieva**, O.I.Romanenko, D.A.Konoplin The molecular aspects of pharmacological activity of naltrexone and naloxone Regional therapy and diagnostics medicinal center "Behterev", 6.Korabelnaja str., Saint Petersburg, 198096, RUSSIA; Phone *-To whom all correspondence should be addressed. Naloxone and naltrexone have been shown to have therapeutic use in medicine. These drugs are a competitive antagonists of opioid receptors. It was demonstrated by mutagenesis studies that several amino acids within the transmembrane domains were important for naloxone and naltrexone binding. By using a site-directed mutagenesis technique it was found that replacement of only a single amino acids residues markedly alters the affinity for antagonists. Some mutations may afford agonist-like properties of antagonists. Chronic administration of opioid antagonists produces increasing opioid receptors density and increasing potency of agonists. This supersensitivity and overdose possibilities after chronic naltrexone treatment in heroin abusers are discussed. Другие интересные материалы:
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
| |||||||||||||||