|
 |
 |
|
|||||||||||||||||||||||
 |
 |
|||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||
|
| |||||||||||||||||||||||||||||||||||||
|



 |
Острые и хронические воздействия опиатами и опиоидами сопровождаются развитием толерантности. Существуют значительные различия в действии агонистов опиоидных рецепторов на рецепторы и сопряженные трансдукторные системы. Наиболее ранними адаптивными процессами при активации рецепторов считаются их десенситизация, понижение плотности (down regulation), интернализация и рециклизация. С их помощью предупреждаются дизрегуляция трансдукторных систем и экспрессии генов. Морфин и другие агонисты, не обеспечивающие десенситизацию, интернализацию и рециклизацию, обладают повышенным потенциалом в плане формирования толерантности. Подобные представления требуют пересмотра ряда положений о механизмах клеточной толерантности. А. Головко, Л. Леонтьева, С. Головко, О. Романенко, Д. Коноплин А. Головко, Л. Леонтьева, С. Головко, О. Романенко, Д. Коноплин Механизмы клеточной толерантности к опиатам и опиоидам
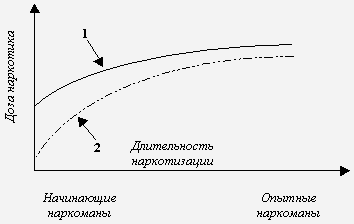
Опиаты и опиоиды относятся к агонистам опиоидных рецепторов. Алкалоиды опийного мака Papaver somniferum морфин, кодеин, тебаин, орипавин и др. носят название опиаты. При ацетилировании алкалоидов образуются полусинтетические опиаты диацетилморфин (героин), 6-моноацетилморфин, ацетилкодеин. Агонисты с иной структурой (пептиды, гетероциклические соединения) относятся к опиоидам. Среди них выделяют синтетические производные (дериваты фентанила, метадон, трамадол и др.) и нейропептиды (эндорфины, энкефалины и динорфины). Термин “опиоидные рецепторы” происходит от названия их естественных агонистов опиоидов-нейропептидов. При длительных воздействиях рассматриваемыми агентами формируется состояние толерантности, под которым понимают ослабление анальгезирующего действия опиатов/опиоидов, их эйфоригенных и седативных эффектов, влияния на ряд физиологических процессов, в особенности на регуляцию температуры тела и функции внешнего дыхания. Термины “толерантность” и “чувствительность” имеют противоположное значение. Толерантность (лат. tolerantia способность переносить, терпеливость; син. переносимость) может определяться как способность организма переносить воздействие определенного лекарственного вещества или яда без развития соответствующего терапевтического или токсического эффекта. Чувствительность - способность организма, его систем, органов или тканей реагировать на воздействие данного вещества [3]. В контексте опиатной наркомании толерантность означает снижение реакций целостного организма в ответ на повторные введения опиатов/опиоидов. Результатом развития толерантности становится постепенное повышение дозы наркотика с целью достижения необходимого эффекта. Например, при формировании зависимости от опиоидов суточная доза может возрастать в десятки раз [2]. Выяснилось, что скорость развития толерантности во многом определяется характером сопутствующего расстройства личности, и что в процессе хронической наркотизации толерантность претерпевает значительные изменения [2]. Например, устойчивость к токсическому действию опиатов/опиоидов после детоксикации или на фоне ремиссии может значительно понижаться. Длительная, многолетняя наркотизация часто сопровождается постепенным снижением толерантности. Это объясняет, почему смертельные передозировки героином чаще происходят у взрослых наркоманов с длительным стажем болезни [56, 59]. Скорость развития толерантности к различным эффектам героина неодинакова. Устойчивость к эйфоригенному и антиноцицептивному действию наркотика возрастает быстрее в сравнении с ростом толерантности к угнетению дыхания. Подобное несоответствие можно представить и в виде кривых прироста значений токсифицирующих доз (т. е. доз, вызывающих необходимый наркогенный эффект) и летальных доз опиатов/опиоидов [56, 59] (рис.1). Видно, что в процессе наркотизации снижается разность между величинами рассматриваемых доз. Результатом сближения двух показателей становится возрастание опасности передозировки у лиц с длительным стажем болезни. Причинами формирования толерантности являются изменения мишеней опиатов/опиоидов - в первую очередь опиоидергической нейротрансмиссии и сопряженных нейромедиаторных систем. Речь идет о многоуровневых перестройках синтеза, накопления и экзоцитоза нейромедиаторов, нейромодуляторов и нейрогормонов, взаимодействующих с ними рецепторов, систем трансдукции и генов. Подобные сдвиги нередко объединяют в понятие “механизм клеточной толерантности” [59]. Опиоидергические механизмы толерантностиКак известно, хроническая наркотизация сопровождается нарушениями синаптической передачи многих нейромедиаторных систем, в первую очередь - опиоидергической. Накоплен достаточный объем информации о синтезе, экзоцитозе и деградации опиоидных нейропептидов в подобных условиях. Не менее подробно изучены процессы передачи синаптического сигнала, включая уровни внутриклеточных трансдукторных систем и генома [53]. Изменениям опиоидных рецепторов в период формирования толерантности уделяется наибольшее внимание. Как известно, длительное воздействие агонистом сопровождается снижением числа рецепторов-мишеней - down regulation. Исследования ex vivo при хронической наркотизации в этом отношении чаще дают противоречивые результаты. В ряде работ установлен факт снижения плотности опиоидных рецепторов после длительной экспозиции к агонистам (down regulation) [44, 47], в других же публикациях приводится противоположный эффект - повышение числа рецепторов (up regulation) [37, 61]. Наиболее устойчиво феномен down regulation достигается при хроническом воздействии опиатами/опиоидами в опытах in vitro [41, 45]. Столь же неоднозначно мнение о том, какой именно тип опиоидных рецепторов (мю, дельта, каппа) вовлечен в формирование толерантности. Вероятно, вовлечены все три типа рецепторов, но наиболее изучены в этом плане мю- и дельта-рецепторы [33, 52, 64]. В качестве доказательства приводятся результаты оценки формирования толерантности у животных, лишенных гена того или иного рецептора. Так, у мутантных мышей без гена дельта-1 и дельта-2 рецепторов не развивалась толерантность к анальгетическому действию морфина [64]. Роль каппа рецепторов в данном явлении менее ясна. Известна способность агонистов каппа рецепторов ослаблять толерантность к морфину. Предполагается, что этот эффект опосредован N-метил-D-аспартатными рецепторами [43]. Общепризнанно, что длительное воздействие опиоидными агонистами снижает чувствительность рецепторов к последующей экспозиции (десенситизация). В основе данного явления лежит фосфорилирование аминокислотных остатков серина и/или треонина в области третьей внутриклеточной петли и внутриклеточной карбоксильной терминали (СОО-терминаль или С-терминаль). С-терминаль, по-видимому, имеет решающее значение в рассматриваемом явлении (рис. 2). Фосфорилирование катализируют протеинкиназы, относящиеся к системам вторичных мессенджеров (Са2+ - кальмодулинзависимая протеинкиназа, протеинкиназы А и С), а так же киназы, связанные с G-белок - рецепторным комплексом (GRKs - G-protein receptor kinases), тирозинкиназа и др. [20, 60]. Существует избирательность протеинкиназ в отношении фосфорилируемых аминокислотных остатков. Например, остаток серина-266 третьей внутриклеточной петли мю-рецептора крысы является мишенью для Са2+ - кальмодулинзависимой протеинкиназы II (в мю-рецепторе человека соответствующий серин находится в положении “268”) [24]. Треонин-353 СОО-терминали дельта-рецептора и серин-375 мю-рецептора - для GRK [14]. Наибольшее число мишеней протеинкиназ расположено в области СОО-терминали. В дельта-рецепторе мыши внутриклеточный С-хвост содержит семь остатков серина и треонина: серин-344 и 363; треонин-335, 352, 353, 358 и 361. При активации рецепторов, экспрессированных в клетках НЕК-293 (human embryonic kidney), агонистом DPDPE ([D-Pen2,Pen5]enkephalin) быстрее всего фосфорилировались серин-363 и треонин-358. Треонин-361 скорее всего выполняет роль места связывания какой-то киназы и сам не подвергается фосфорилированию [20]. СОО-терминаль мю-рецептора крысы на участке “треонин-354 - треонин-394” содержит 12 остатков треонина и серина, рассматриваемых в качестве возможных мест для фосфорилирования. Наиболее “активная” зона - от серина-363 до серина-375 [18]. Фосфорилированный рецептор связывается с белком аррестином, отщепляется от G-белка и подвергается интернализации (эндоцитоз, секвестрация) в особые углубления клеточной мембраны, покрытые белком клатрином [26]. Из них формируются первичные (ранние) эндосомы, в кислой внутренней среде которых и осуществляется восстановление исходного состояния опиоидного рецептора посредством дефосфорилирования и отщепления лиганда. Завершается процесс встраиванием рецептора в клеточную мембрану (рециклизация) и восстановлением его функциональной активности (ресенситизация). Часть иммобилизованных в первичных эндосомах рецепторов может транспортироваться в лизосомы для конечной деградации [51]. Целесообразно разграничивать термины, относящиеся к данной проблеме. Так, явление down regulation можно обозначить как уменьшение плотности нейрорецепторов на пре- и постсинаптической мембране. Методически такое состояние можно выявить с помощью радиолигандного анализа или радиоаутографии. Интернализация - это процесс перемещения рецепторной молекулы от цитоплазматической мембраны в цитозоль. Визуализация интернализации достигается с помощью иммуноцитохимических методов. Интернализация - наиболее характерная реакция мю- и дельта-рецепторов в ответ на воздействие агонистом. Для каппа-рецепторов подобная реакция менее характерна. Так, эторфин, неселективный агонист для опиоидных рецепторов всех типов, вызывал быструю интернализацию дельта-рецепторов мыши, клонированных в клетках НЕК-293. На каппа-рецепторах такие эффекты не выявлялись даже при насыщающих концентрациях агониста [12]. Десенситизация - ослабление опиоидергической нейропередачи. Поведенческими эквивалентами десенситизации можно было бы считать снижение анальгетической, эйфоригенной и иной фармакологической активности опиатов/опиоидов, что, по сути дела, аналогично рассмотренным выше признакам толерантности. Нейрохимическим коррелятом десенситизации является ослабление эффектов опиоидных агонистов в отношении регулируемых рецепторами трансдукторных систем (циклических нуклеотидов, ионных каналов и др.) [10, 14, 60] с последующей редукцией клеточного ответа [55]. Все же начальным звеном данного процесса является фосфорилирование и нарушение взаимодействия рецептора с G-белком (uncoupling), которое, в свою очередь, инициируется присоединением к рецептору белка аррестина [57]. Не вполне корректно считать снижение плотности рецепторов (down regulation) и интернализацию эквивалентами десенситизации. Уместнее говорить о том, что десенситизацию могут сопровождать оба явления, хотя по времени эти события чаще не совпадают [27, 28]. Эксперименты на клонированных опиоидных рецепторах показывают, что развитие десенситизации зависит от многих факторов: используемой клеточной системы, химической структуры агонистов, длительности инкубации, свойств самого рецептора, в том числе характера мутации и т.д. Так, морфин и леворфанол, обладающие высоким аддиктивным потенциалом, не десенситизировали мю-рецепторы, клонированные в клетках НЕК-293. В сходных условиях метадон и бупренорфин вызывали десенситизацию. Авторы связывают данный феномен с клинической эффективностью метадона и бупренорфина, используемыми в терапии опиатной наркомании [7]. Взаимосвязь процессов фосфорилирования, десенситизации, снижения плотности и интернализации опиоидных рецепторовВ работах [55, 63] предпринята попытка провести связи между процессами фосфорилирования и интернализации. Были использованы мутантные дельта-рецепторы, лишенные аминокислотных остатков - предполагаемых мишеней для протеинкиназ. Воздействие агонистами на такие рецепторы не сопровождалось их интернализацией. Высказано предположение о возможной связи между фосфорилированием возбужденного агонистом рецептора и его интернализацией. Следует уточнить, что в работах [55, 63] дельта-рецепторы экспрессировали в клетках СНО (Chinese hamster ovary) и в клетках нейробластомы-глиомы NG108-15. В процессе дальнейшей разработки проблемы стали появляться уточняющие, а иногда противоречивые факты. Так, S.M.Murray и др. [36] установили, что интернализации дельта-рецептора необязательно должно предшествовать фосфорилирование. В их исследовании дельта-рецепторы мышей экспрессировали в клетках НЕК-293. Были созданы мутантные формы, лишенные возможных участков фосфорилирования в области СОО-терминали. Воздействие агонистами эторфином и DADLE ([D-Ala2,D-Leu5]enkephalin) сопровождалось интернализацией как рецепторов “дикого типа”, так и мутантов. Это позволило авторам усомниться в решающей роли фосфорилирования для последующей интернализации. Кроме того, по их мнению, при интерпретации подобных результатов следует учитывать характеристики клеточной системы, в которой экспрессируются рецепторы. Y.Pak и др. [42] выяснили, что снижение плотности мю-рецепторов, вызываемая агонистом DAMGO ([D-Ala2,N-methyl-Phe4,Gly5-ol]enkephalin), регулируется двумя различными путями клеточной трансдукции. Первый сопряжен с G-белком и связанных с ним киназами (GRKs). Второй путь - G-белок-независимый. В нем решающую роль играет фосфорилирование рецептора тирозинкиназой. На мю-рецепторах крысы (клонированы в клетках НЕК-293) продемонстрировано базальное (т.е. в отсутствии агониста) фосфорилирование серина-363 и треонина-370. Агонист DAMGO индуцировал фосфорилирование остатка треонина в положении 370 и серина-375. Мутация Серин 375 Аланин* сопровождалась ослаблением скорости и величины интернализации рецепторов под действием DAMGO, а мутации Серин 363 Аланин и Треонин 370 Аланин приводили к противоположным эффектам. Это означает, что фосфорилирование может не только инициировать эндоцитоз рецепторов, но и противодействовать ему. В данном исследовании установлена интернализация после выключения из процессов фосфорилирования всех трех аминокислот (мутация с заменой на аланин). Следовательно, подтвержден факт о вовлечении в интернализацию не только фосфорилирования, но и иных механизмов [8]. Предполагается, что интернализации предшествует процесс перехода димерной формы опиоидного рецептора в мономерную. Установлено, что экспрессированные в клетках СНО дельта-рецепторы находятся преимущественно в форме димеров. При внесении в инкубационную среду агонистов DADLE, DSLET ([D-Ser2]Leu-enkephalin-Thr), DPDPE и эторфина отмечался процесс мономеризации димеров, который предшествовал интернализации рецепторов. У морфина такие эффекты не выявлены. В этом исследовании показана решающая роль концевого участка из 15 аминокислот СОО-терминали в процессах мономеризации и интернализации дельта-рецептора [15]. Снижение содержания димеров мю-рецепторов мышей (экспрессированы в клетках НЕК-293) наблюдалось при воздействии агонистом DAMGO [25]. Существует мнение, что опиоидные рецепторы различных классов способны формировать гетеродимеры. Такие данные получены в отношении мю- и дельта-рецепторов [49, 50]. При хронических воздействиях морфином выявляется избирательное возрастание плотности дельта-рецепторов, что изменяет состояние комплекса “мю-дельта”, приводя в конечном итоге к нарушению опиоидергической нейропередачи в процессе развития толерантности [48, 49]. Имеются определенные сложности при попытке провести параллели между толерантностью, процессами десенситизации и интернализации и внутримолекулярными событиями, следующими за связыванием агониста с рецептором (активация G-белка и систем вторичных мессенджеров, фосфорилирование). Дело в том, что информация по данной проблеме накапливается преимущественно за счет экспериментов in vitro, в которых мишенями агонистов служат клонированные рецепторы. Можно предположить, что экстраполяции полученных результатов на более сложные биологические системы весьма проблематичны. Клонирование опиоидных рецепторов, лишенных части СОО-терминали, а также мутации отдельных аминокислотных остатков молекулы рецептора изменяют скорость развития десенситизации и интернализации. Воздействие агонистом DADLE на дельта-рецептор, лишенный семи последних аминокислот, сопровождалось быстрой интернализацией. В то же время мутанты без 15 или 37 аминокислотных остатков в сходных условиях эксперимента подвергались медленной интернализации. Было высказано предположение о решающей роли остатков серина и треонина между треонином-335 и глицином-365 внутриклеточного хвоста. Точечные мутации Серин 344 Глицин, Треонин 352 Аланин, Треонин 353 Аланин, Треонин 358 Аланин и Серин 363 Аланин полностью подтвердили такое предположение [55]. Замена треонина-394 СОО-терминали мю-рецептора крысы на аланин тормозила десенситизацию. Но одновременно выяснилось, что мутантные рецепторы Треонин 394 Аланин в процессе длительного воздействия агонистом DAMGO значительно быстрее подвергались явлению down regulation, интернализации и ресенситизации в сравнении с “диким типом” [60]. V.Segredo и др. [51] показали, что мутантный мю-рецептор, лишенный карбоксильного хвоста от треонина-354, подвергается интернализации даже без предшествующего воздействия агонистом. В дельта-рецепторе одной из таких “ключевых” аминокислот является треонин-353, так же относящейся к СОО-терминали. Мутация Треонин 353 Аланин полностью предупреждала интернализацию после воздействия агонистом DADLE [14]. В этом же исследовании установлено, что серин-344, являющийся мишенью протеинкиназы С, не принимает участия в развитии интернализации. Вместе с тем, роль протеинкиназы С в инициации процессов понижения плотности и интернализации дельта-рецепторов доказана ранее [19]. По-видимому, протеинкиназа С имеет несколько мишеней в области терминального хвоста, каждая из которых играет строго определенную роль. Появляется все больше доказательств того, что имеет место относительная самостоятельность процессов фосфорилирования, десенситизации, уменьшения плотности, интернализации и ресенситизации. N.Trapaidze и др. [55] не обнаружили достоверного влияния модуляторов протеинкиназ А и С, а также фосфатаз на величину базальной интернализации дельта-рецепторов мышей (так называемая конституциональная интернализация - в отсутствии агониста). Сходный результат получен и в присутствии DADLE. В работе [11] десенситизацию мю-рецепторов (клонированы в клетках СНО) вызывали даже после замены всех остатков треонина и серина в третьей внутриклеточной петле и в СОО-терминали. Достаточно независимы и два тесно связанных явления как down regulation - исчезновение рецепторов с поверхности клеточной мембраны и интернализация - перемещение гликопротеиновой молекулы внутрь клетки [55]. То же можно сказать и в отношении влияния G-белков на понижение плотности рецепторов и их интернализацию. Активация G-белков - необходимое условие при опиоидергической передаче. Однако выяснилось, что развитие явления down regulation происходит как при возбуждении G-белка, так и без такового [42]. Одно из возможных объяснений перечисленных выше противоречий - параллельное протекание описываемых событий. К примеру, медленная десенситизация, не совпадающая по времени с фосфорилированием, понижением плотности рецепторов и эндоцитозом, может быть объяснена одновременным восстановлением активного пула рецепторов посредством рециклизации, то есть за счет возвращения рецепторов из цитозоля в клеточную мембрану. В исследовании [27] мю-рецепторы клонировали в клетках SHSY5Y нейробластомы человека. Агонист DAMGO инициировал быстрое фосфорилирование рецепторов, тогда как развитие десенситизации запаздывало. Однако это несоответствие исчезало после блокады процессов рециклизации. Уникальность мю- и дельта-рецепторов состоит в том, что на некоторых клеточных моделях агонисты с различной структурой инициируют неодинаковые реакции. Воздействие пептидами DAMGO (мю-агонист), DADLE (дельта-агонист) и морфиноподобным соединением эторфином (неселективный мю-, дельта-агонист) сопровождалось быстрым снижением плотности рецепторов, экспрессированных в клетках НЕК-293. Морфин таким эффектом не обладал [4, 23, 51]. Морфин так же не инициировал феномен down regulation дельта-рецепторов в другой клеточной системе - нейробластоме-глиоме NG108-15 [19], а по способности вызывать интернализацию мю-рецепторов значительно уступал DAMGO и метадону (эксперименты на нейронах гиппокампа крыс) [10]. Толерантность, вызываемая морфиномКакие же особенности рассмотренных этапов синаптической передачи могут иметь отношение к толерантности, вызываемой агонистами опиоидных рецепторов? В первую очередь толерантности, вызываемой основным метаболитом героина морфином. Существуют особенности взаимодействия агонистов пептидной природы с зоной связывания опиоидного рецептора в сравнении с алкалоидами, в том числе морфином. Зону рецептирования лигандов опиоидного рецептора условно делят на участки селективности и карман связывания. Первые расположены преимущественно выше наружной поверхности мембраны и сформированы аминокислотными остатками внеклеточных петель и верхушек трансмембранных доменов [32, 35]. Карман связывания находится ниже наружной поверхности мембраны. Его образуют спиральные петли трансмембранных доменов [5, 6]. Лиганды-пептиды взаимодействуют как с участками селективности, так и с карманом связывания. Алкалоиды (морфин, эторфин) и антагонисты налоксон и налтрексон связываются преимущественно в области кармана. При этом заряженный азот молекулы лиганда способен взаимодействовать с остатками ароматических аминокислот полипептидной цепи [5, 6, 29, 32]. Очевидная закономерность: пептидные агонисты, уступающие морфину по способности инициировать толерантность, имеют особые места специфического связывания. Но, с другой стороны, эторфин взаимодействует с аналогичным для морфина участком рецептора. Однако формирование толерантности при воздействиях эторфином происходит медленно [17]. Связывание морфина сопровождается угнетением аденилатциклазы с вовлечением G-белка. Подобный механизм свойственен и другим опиоидным агонистам [57]. Но, в отличие от пептидных агонистов и эторфина, морфин слабо активизирует процессы фосфорилирования опиоидных рецепторов [25, 62]. Предполагается, что морфин стабилизирует мю-рецептор в конформации, при которой становится невозможным фосфорилирование с участием протеинкиназ, связанных с G-белок - рецепторным комплексом (GRKs) [25, 58, 62]. Возможно, что существуют несколько конформаций возбужденного опиоидного рецептора в зависимости от структуры агониста. Характер конформационной перестройки предопределяет возможность и эффективность взаимодействия рецептора с протеинкиназами [62]. В опытах in vitro посредством избыточной экспрессии (overexpression) протеинкиназы, связанной с G-белок - рецепторным комплексом, удавалось усилить способность морфина инициировать процессы фосфорилирования и интернализации мю-рецепторов [57, 62]. В опытах ex vivo выявлено возрастание экспрессии белка протеинкиназы GRK в голубом пятне крыс на фоне хронической морфинизации. Данный факт можно рассматривать как адаптивную реакцию нейронов на избыточное воздействие наркотиком [54]. Подобным же образом можно объяснить и увеличение содержания киназы GRK2 во фронтальной коре героиновых наркоманов, умерших от передозировки [40]. Последующие события, инициируемые морфином, также значительно отличаются: он не вызывает выраженной десенситизации, понижения плотности и интернализации опиоидных рецепторов [4, 23, 51]. Анализ изложенного позволяет утверждать, что уже на самых ранних этапах опиоидергической нейротрансмиссии морфин действует иначе в сравнении с другими агонистами. Подобные различия создают условия для возникновения толерантности. Постепенно сформировалось мнение о возможности влияния на процесс связывания наркотика с рецептором и о возможности изменения хода последующих событий. Первым шагом в этом направлении стала оценка толерантности в режиме совместного введения морфина и антагонистов опиоидных рецепторов налоксона, налтрексона и налмефена. При таких условиях достоверно снижалась скорость возникновения толерантности к наркотику (оценивалась по анальгетическому действию). Следовательно, теоретические разработки стали предпосылкой для развития нового клинического направления - оптимизации использования морфина для лечения болевого синдрома [13]. Активация морфином опиоидного рецептора не сопровождается связыванием рецептора с белком аррестином для последующей секвестрации внутрь клетки [62]. Скорее всего, подобное явление является следствием неспособности наркотика инициировать фосфорилирование. В качестве доказательства приведем результаты работы [62]. В клетках НЕК-293 совместно экспрессировали мю-рецепторы, протеинкиназу, связанную с G-белок - рецепторным комплексом (GRK) и аррестин. Морфин вызывал интернализацию рецепторов при избыточной экспрессии протеинкиназы и аррестина (его содержание в клетке возрастало в 30 и более раз). Этот эффект исчезал, если блокировался этап фосфорилирования. Для десенситизации опиоидных рецепторов после экспозиции к агонисту необходима диссоциация комплекса “рецептор - G-белок” [9]. Если абстрагироваться от ряда противоречий при описании событий вслед за возбуждением опиоидного рецептора (см. выше) и следовать “классическим” представлениям, можно говорить, что диссоциация нервного окончания и G-белка возможна только при фосфорилировании рецептора и присоединении к нему белка аррестина. Как уже обсуждено ранее, эти процессы не характерны для действия морфина. При избыточной экспрессии протеинкиназы GRK и аррестина удавалось добиться десенситизации мю-рецептора с помощью наркотика [57]. Роль бета-аррестина-2 в формировании толерантности к анальгетическому действию морфина оценивалось на мышах, лишенных гена названного белка. Животные получали наркотик в дозе 10 мг/кг массы тела в сутки на протяжении 9 дней. С помощью теста горячей пластины было констатировано отсутствие изменений анальгетического действия опиата. В контрольной группе мышей (дикий тип) в процессе наркотизации прогрессивно развивалась толерантность. В параллельной серии экспериментов оценивалось влияние мю-агониста DAMGO на специфическое связывание 35S-гуанозинтрифосфата с синаптическими мембранами ствола головного мозга грызунов опытной и контрольной групп (данный подход позволил судить о состоянии системы “опиоидный рецептор - G-белок”). К исходу 5-х суток у контрольных мышей агонист не влиял на параметры связывание 35S-гуанозинтрифосфата (развитие десенситизации). У мышей-мутантов параметры радиолигандного исследования под воздействием хронической наркотизации не изменялись в сравнении с исходными данными (отсутствие десенситизации). Таким образом роль бета-аррестина-2 в формировании десенситизации и толерантности при хроническом воздействии морфином доказана в опытах in vitro и ex vivo [8]. Внутриклеточный карбоксильный хвост мю-рецептора играет решающую роль в торможении процессов фосфорилирования, десенситизации, интернализации и ресенситизации, наблюдаемых при воздействии морфином. Наиболее значимый участок - финальная аминокислотная последовательность, начиная от лейцина-387. Были созданы сплайс-варианты, резко отличавшиеся от рецептора “дикого” типа именно по полипептидной цепи от позиции “387”. При воздействии морфином на некоторые из таких мутантов выявлялись фосфорилирование, десенситизация, интернализация и ресенситизация, чего не наблюдалось при активации наркотиком рецепторов “дикого” типа [25]. Возникает парадоксальная ситуация: высокоаффинные агонисты, обладающие способностью активировать опиоидный рецептор, запускать процессы фосфорилирования, десенситизации, понижения плотности, интернализации и ресенситизации, значительно уступают морфину в плане формирования толерантности [58]. Клетка включает адаптационные механизмы (десенситизацию и интернализацию) для предупреждения дальнейшего действия агониста [9]. В таком случае снижается вероятность модуляции ряда внутриклеточных биохимических путей, в том числе и экспрессии генов. Морфин в этом отношении может действовать более длительно, поскольку защитные механизмы клетки рецепторного уровня срабатывают менее эффективно. Появляется возможность для глубоких изменений структурно-метаболических комплексов клетки (обеспечивающих энергообмен, пластические процессы, функцию мембран и ионных каналов, хранение генетической информации и др.). Следовательно, развитие толерантности под влиянием опиоидных пептидов, эторфина и других агонистов можно представить в виде процесса, направленного извне в клетку, в то время как морфин индуцирует толерантность, идущую как бы изнутри, от генома. Если первый вид устойчивости более или менее ясен и структурирован (активация опиоидного рецептора ® фосфорилирование ® десенситизация ® интернализация), то второй гораздо сложнее и включает модуляцию не только опиоидергических систем (как прямым образом, так и посредством изменения экспрессии генов), но и иных нейромедиаторов, многих биохимических процессов в клетке. Эти сдвиги могут составлять нейрохимическую основу толерантности, вызываемую морфином [10, 22]. Предполагается следующая цепь событий. Активация опиоидных рецепторов индуцирует каскады внутриклеточных трансдукторов (циклазные, фосфоинозитидный, кальциевый и др.). Вторичные мессенджеры через систему протеинкиназ воздействуют на транскрипцию “ранних” генов - c-fos, fos-B, c-Jun, Jun-B, Jun-D, Fra-1, Fra-2, Krox-20, Krox-24 и др. [1, 21]. Экспрессируются белки Fos, Jun, CREB и др., которые способны формировать гомо- и гетеродимеры. Димеры связываются со специфическими участками ДНК в промоторных областях многих “поздних” генов и регулируют их транскрипцию. Опиаты/опиоиды сопряжены с “ранними” генами преимущественно через циклазную систему. Так, циклический аденозинмонофосфат влияет на ДНК через “ранний” ген CREB и соответствующий белок ssCRE-BP (single-stranded cyclic AMP response element-binding protein). Длительная наркотизация опиатами/опиоидами изменяет активность названной системы [16, 31, 39]. Показано, что ядерный фактор ssCRE-BP представляет собой полипептид с молекулярной массой 110-150 kDa. Он включает два домена, содержащих значительное число остатков глицина и глутамина. Выделена также мРНК, кодирующая изученный белок [38, 39]. Подобные теоретические рассуждения не выходили бы за рамки обычной научной дискуссии, если бы не были известны два неблагоприятных побочных свойства анальгетика морфина: высокий аддиктивный потенциал и способность быстро формировать толерантность. Если предположить, что развитие десенситизации и интернализации рецепторов - нормальная адаптивная реакция клетки при избыточном действии опиоидных агонистов [57], то следующие за перечисленными событиями рециклизация и ресенситизация опиоидных нервных окончаний возвращают клетку в исходное состояние. Кроме того, блокирование трансдукторного сигнала, вызванного возбуждением опиоидного рецептора, уменьшает вероятность развития последующих внутриклеточных реакций. В первую очередь это относится к удержанию в состоянии равновесия основных структурно-метаболических комплексов клетки, в том числе и экспрессии генов. Морфин, способный возбуждать опиоидные рецепторы, активизировать системы вторичных передатчиков, не формирует “обратную связь” для предупреждения избыточного действия на клетку-мишень. Процессы фосфорилирования, десенситизации и интернализации замедлены. Отсюда вытекает, что действие наркотика будет более “глубоким” и может достигать уровня экспрессии генов. Толерантность в таком случае могут определять не только изменения различных звеньев опиоидергических систем, но и серьезные сдвиги сопряженных нейромедиаторов, ионных каналов, клеточных мембран, пластических процессов, энергообмена и др. Непосредственным инициатором перечисленных отклонений становится не избыточная активация агонистом рецепторов, а нарушения экспрессии генов. Таким образом, при длительном воздействии опиатами/опиоидами развиваются процессы десенситизации и интернализации опиоидных рецепторов. Подобная реакция может рассматриваться в рамках адаптационной перестройки, защищающей клетку от чрезмерного воздействия фармакологическим агентом. При действии морфина десенситизация и интернализация замедленны, что создает условия для избыточной активации трансдукторных систем и экспрессии генов. Нарушения на уровне транскрипции могут составлять основу толерантности при хронических экспозициях к наркотикам. Рис. 1. Кривые прироста летальных (1) и эйфоригенных (2) доз опиатов/опиоидов в процессе наркотизации [56, 59].
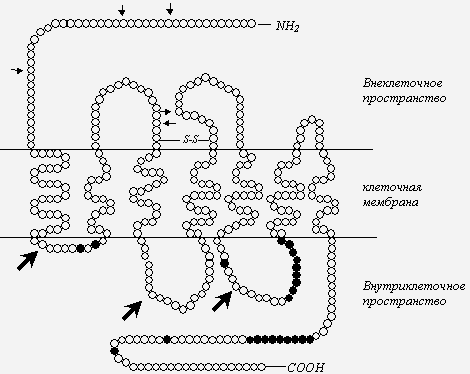
Рис. 2. Схема опиоидного рецептора [34, 46], с дополнениями.
Примечания: темными кружками обозначены аминокислоты, подвергающиеся фосфорилированию посредством протеинкиназ; стрелки - места возможного гликозилирования; Литература
Сведения об авторах Головко Александр Иванович*. Заведующий стационарным отделением регионального лечебно-диагностического медицинского центра "Бехтерев", доктор медицинских наук профессор. 195027, Санкт-Петербург, Большеохтинский пр., дом 31, кв.5; д.т. 227-20-22, (отв. за переписку), сл. тел. 183-93-57, 183-92-75, 324-04-18, E-mail: prgolovko@mail.ru Леонтьева Любовь Владимировна. Ассистент-исследователь Университета шт.Западная Виржиния, Box 9151, Morgantown, WV 26505-9151, U.S.A., тел. (304) 293-15-28, факс (304) 293-02-65, E-mail: Luba 105@hotmail.com Головко Cергей Иванович. Старший научный сотрудник Центра, кандидат медицинских наук. 197348, Санкт-Петербург, Богатырский пр., дом 4, кв.724; д.т. -393-11-18, сл. тел. 183-93-57, 183-92-75, 324-04-18, 393-85-09. Романенко Олег Иванович. Ординатор стационарного отделения Центра, кандидат медицинских наук. 198005, Санкт-Петербург, 4-я Красноармейская ул., дом 2/35, кв. 45; д.т. 316-40-79, сл. тел. 183-93-57, 183-92-75, 324-04-18. Коноплин Дмитрий Алексеевич. Ординатор стационарного отделения Центра. 193177, Санкт-Петербург, ул. Караваевская, дом 22, кв. 15; д.т. 107-39-42, сл. тел. 183-93-57, 183-92-75, 324-04-18. A.I.Golovko1, L.V.Leontieva2, S.I.Golovko1, O.I.Romanenko1, D.A.Konoplin1 MECHANISMS OF CELLULAR TOLERANCE TO OPIATES AND OPIOIDS 1Regional Therapy and Diagnostics Medicinal Center “Behterev”, Ulitsa Korabelnaja, 6, Saint Petersburg, 198096, RUSSIA; fax (812) 321-1324, E-mail: prgolovko@mail.ru; 2West Virginia University, Box 9151, Morgantown, WV 26505-9151, USA; fax (304) 293-0265, E-mail: Luba 105@hotmail.com. Acute and chronic opiates/opioids treatment results in drug tolerance. Individual agonists may have substantially different effects on the regulation of opioid receptors and transduction systems. Receptor desensitization, down regulation, internalization and recyclization are the early adaptive processes. They are a protective mechanisms whereby cells avoid the disregulation of transduction pathways and gene transcription. Those agonists that do not cause receptor desensitization, internalization and recyclization such as morphine may have higher potencies to induce tolerance. That view may be as fundamental revision of our understanding of the role of opioid receptors desensitization endocytosis and recycling in the biology of tolerance. Другие интересные материалы:
| |||||||||||||||||||||||||||||||||||||||||||||
|
| |||||||||||||||